慶應発サイエンス
single cell RNA-seqデータの普遍的な特徴を抽出することに成功 -疾患解析データの本質を捉えることが可能に-
研究の背景
一つの細胞ごとに遺伝子発現状態が分かる技術とは
一細胞生物学(シングルセル・バイオロジー)の技術は生物学・医学研究において観測可能な情報の質と量を劇的に改善してきました。中でもsingle cell RNA-sequence(scRNA-seq)は遺伝子発現の状態を一細胞レベルで観察することができます。RNA-seqデータは探索的研究から仮説検証に至るまで様々な分野の様々な用途で分析対象となっていますが、サンプルの細胞種が不明な状態でデータが得られるため、データから細胞種を推定することが必要となります。このように得られた情報から細胞種を推定し、各サンプルに情報を割り当てることをアノテーションと言います。
これまで行われてきたscRNA-seqにおける解析手法の問題点
従来のアノテーション方法では、複数の細胞集団の間で発現している遺伝子の違いを網羅的に探索する手法(発現変動遺伝子解析)によって、サンプル間での相対的な比較を行い、有意に発現量が上昇している遺伝子群から細胞種を推定します。しかし、遺伝子発現量の数値は、サンプルの特性や定量手法、データ処理方法などの影響を受けて変動するので、細胞種の違い以外にも、サンプル特性や個体差の影響を強く受けてしまうというデメリットがありました。
また、そもそも細胞種の概念は古典的に形態と機能によって定義されてきたため、形態と機能情報がないscRNA-seq(すり潰した細胞集団からmRNAを読み取って解析するため、細胞の形や機能は分からない)において、細胞種を定義づけるようなマーカー遺伝子に関して、発現量の厳密な基準値は明らかになっていないのです。
これらの観点から、アノテーションの結果が古典的な定義での細胞種として、どの程度妥当であるか可視化することは困難でした。さらに、異なるデータセットにおいて同じ細胞種としてアノテーションされた細胞群がどの程度同一の集団として扱えるのかという点も不透明でした。
研究結果
マーカー遺伝子をネットワークとしてとらえる
上記の課題を克服すべく、本研究では、従来法とは異なったフレームワークで細胞群の特色を評価するための手法と背景理論の構築を行いました。細胞の機能や、それをマーカー遺伝子によって説明した細胞種の古典的な定義をヒントに、遺伝子発現の統計学的従属性をRNA-seqデータでみられる細胞の機能としてとらえ、それらの関係を遺伝子制御ネットワーク(Gene Regulatory Network:GRN)として可視化し、GRNの類似性によって細胞特性の近さを評価する指標を開発しました(図1)。GRNの設計に関して、データには現れないような生物学的な知見を実験者が事前にモデルに組み込むことで比較対象を適切に設定できるように、GRNで扱う遺伝子を自由に選択できるような設計にしました。
さらに、本研究で構築した細胞群の類似性の比較尺度をアノテーションに応用し、先行研究にて取得されたオープンソースのscRNA-seq データに対して実施したところ、従来法よりもデータ横断的な特徴を反映したアノテーションに成功しました。
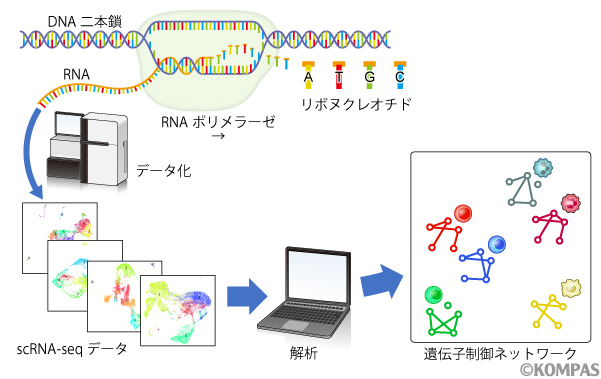
図1.研究の概念図
研究の成果と意義・今後の展開
今回の研究では、細胞種の古典的定義方法とRNA-seqデータ解析における細胞腫の決定方法のミスマッチを指摘しました。また、RNA-seqデータにおいて普遍的に観測可能な細胞機能の特性を、発現量の数値そのものとは異なった方法で表現するための理論構築を行いました。さらに、そのように新たに定式化された細胞特性の表現方法に、古典的細胞種に対して蓄積された生物学的知見を自然に導入する手法を開発しました。
細胞の「普遍的な特性」を表現することは、分析対象がどれだけ自然で一般化された集団であるかを確認するうえで力を発揮するため、モデル動物や培養細胞といった妥当性を事前に検証すべき対象を扱う発生学的研究や病態解析研究などの解析において有用です。
また、RNA-seqデータの時系列的解析や、希少疾患の解析など、複数の個体や複数の研究機関から取得されたデータを統合して解析する必要がある研究テーマにおいて、データに恣意的な加工を施さずに普遍的特性を確認することができるため、様々な分野における応用が期待されます。
今後の展望として、本研究の成果を疾患解析へ応用し、治療ターゲットの発見に役立てていくとともに、学術的には、本研究で集合論的に定式化された細胞集団とその特性の近さについて、位相幾何学などの観点からさらなる考察を行っていくことを検討しています。
特記事項
本研究は、坂口光洋記念慶應義塾大学医学振興基金、日本損害保険協会交通事故医療特定研究助成、武田科学振興財団、JSPS科研費JP22K16696、Keio University Yagami Data Security Labの支援によって行われました。
参考文献
A set-theoretic definition of cell types with an algebraic structure on gene regulatory networks and application in annotation of RNA-seq data.
Okano Y, Kase Y, Okano H.
Stem Cell Reports. 2023 Jan 10;18(1):113-130. doi: 10.1016/j.stemcr.2022.10.015.

左より:岡野栄之(生理学教室教授)、岡野雄士(同教室医学部5年)、加瀬義高(同教室特任講師)
SHARE