病気を知る
血栓性微小血管障害症(thrombotic microangiopathy: TMA)
概要
血栓性微小血管障害症(thrombotic microangiopathy:TMA)は、1)微小血管内に血小板のかたまりが生じ(微小血管内血小板血栓)、2)血小板が消費されて減少し(消費性血小板減少症)、3)閉塞した血管を通過する赤血球が破壊されて貧血(微小血管障害性溶血性貧血)になるという、この3つの特徴をもつ病気の総称です。このように微小血管が障害されることで、脳や消化管や腎臓などに虚血(血流が行き渡らないこと)がおこり、腎臓や脳神経を中心とした多臓器不全がおきます。TMAの範疇に含まれる有名な症候群として、1924年にMoschcowitzによって最初に報告された血栓性血小板減少性紫斑病(thrombotic thrombocytopenic purpura: TTP)と、溶血性尿毒症症候群(hemolytic uremic syndrome: HUS)があります。これらの疾患に典型的な徴候が揃うことは少なく両者を区別することが困難な場合が多いため、TTP/HUSと記載されたり、この2つを包括した病態としてTMAと呼ばれたりします。また、感染症、妊娠、薬剤、高血圧症、臓器移植、造血幹細胞移植、自己免疫疾患、悪性腫瘍に関連してみられることもあります。しっかりとした評価と基礎疾患に応じた治療が大切です。本稿ではTMAに統一してご紹介します。
症状
TMAに特徴的な臨床症状としてとして、血小板減少による紫斑、溶血性貧血による全身倦怠感、動悸、呼吸困難、腎機能障害があります。そのなかで、1)腎機能障害が強く出て尿量が減ったり尿が出なくなったりする場合(=溶血性尿毒症症候群)と、2)腎機能障害は軽度ですがそれに加えて高熱が出たり時間帯によって変動する精神障害(意識障害、気分変動など)が強く出たりする場合(=血栓性血小板減少性紫斑病)があります。これら、微小血管障害性溶血性貧血、血小板減少症、発熱、腎機能障害、動揺する精神神経症状をMoschcowitzの古典的5徴候と言います。ただし、これらの症状が揃うことはむしろ少ないと言え、両者を区別することは難しいことが多いです。現在では、溶血性貧血と血小板減少症の2徴候が揃うことが重要とされています。心臓や肺や消化管に症状がでることもあります。腸管出血性大腸菌が原因となるTMA(=STEC-HUS)の場合には、下痢や血便、腹痛といった腸炎症状が最初に生じ、数日後から上記のような溶血性尿毒症症候群を発症することがあります。
診断
TMAは臨床症状、検査所見、末梢血塗抹標本における破砕赤血球の増加により診断することが原則です。血栓性血小板減少性紫斑病と非典型的溶血性尿毒症症候群については厚生労働省の診断基準も参考にします(詳しくは難病情報センター![]() のページをご参照ください)。
のページをご参照ください)。
血液検査・尿検査では下記の異常をみます。
- 血小板減少(15万/μL未満)
- 溶血性貧血:末梢血中の破砕赤血球の出現、網赤血球数の増加、ヘモグロビン低下(10g/dL未満)、LDH・間接ビリルビン上昇、ハプトグロビン低下、直接クームス試験陰性
- 腎機能障害:クレアチニン上昇、尿素窒素上昇、蛋白尿・血尿出現
- ADAMTS13活性低下、抗ADAMTS13抗体(インヒビター)陽性 (後述)
- 便培養・便中ベロ毒素検査(腸管出血性大腸菌によるTMAの場合)
この中でも特徴的なのは破砕赤血球が出現することです(図1)。
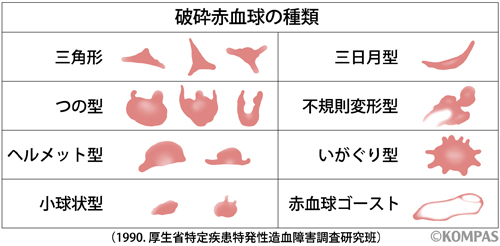
図1.破砕赤血球とその種類
病因による分類
近年、TMAの病因の一部が判明し始めており、多彩な病気を含むTMAを病因毎に分類して説明します。
病因が判明しているTMA
1. 感染症
O157やO111といった腸管出血性大腸菌(shiga toxin-producing escherichia coli:STEC)による腸炎のあとに発症するTMA(=STEC-HUS)が、TMAの原因として最多と言えます。腸管出血性大腸菌がTMAを発症させるメカニズムには分かっていないことも多いのですが、この菌の作るベロ毒素(shiga toxin)が中心的な役割をもっていると推測されています。牛肉や生野菜を含む食事歴や井戸水の摂取歴が重要になります。腸管出血性大腸菌による腸炎にかかり下痢や発熱が出てから4~10日後に血小板減少、細血管障害性溶血性貧血、腎機能障害を来します(=溶血性尿毒症症候群の3徴)。溶血性尿毒症症候群の90%は腸管出血性大腸菌によるものといわれています。我が国での2019年の統計では3,744人の腸管出血性大腸菌感染者のうち溶血性尿毒症症候群を発症したのは78人と報告されています。
その他、肺炎球菌によってTMAが発症する報告もありますが、これは極めてまれです。肺炎球菌が産生するノイラミニダーゼが原因と考えられています。
2. 補体制御異常
血小板減少、溶血性貧血、腎機能障害という溶血性尿毒症症候群に特徴的な症状を持ちながら、上記のような腸炎の症状がない患者さんがまれにいます。これを非典型溶血性尿毒症症候群(atypical HUS:aHUS)または補体介在性TMA(complement-mediated TMA:CM-TMA)と呼びます。最近、原因の解明が進み、生体防御反応に関わる補体という分子に対する制御機構の先天的な遺伝子異常が報告されつつあります。遺伝子異常で最も多いのがH因子の異常、ついで膜補因子蛋白(MCP)と報告されています。また、H因子に対する後天的な自己抗体産生も原因となる場合があります。これらの遺伝子異常や自己抗体により補体が異常に活性化され、本来は攻撃されないはずの微小血管内皮細胞(血管の壁を形作る細胞)に障害をきたすことで、TMAを発症するといわれています。感染症、分娩、手術など補体系が急激に活性化される原因をきっかけに発症することが知られており、日本では約7割の症例で何かしらのきっかけが確認されています。補体の異常な活性化は一般的な血液検査で評価することは困難ですので、専門的な遺伝子検査や抗H因子抗体の検査が必要になります。腸炎に伴う溶血性尿毒症症候群とは異なり、非常に難治性の病気です。先天性でも後天性でも中等症以上であれば医療費助成の対象になっています。
3. ADAMTS13の異常
ADAMTS13はメタロプロテアーゼという一群に属する蛋白切断酵素で、止血に関わるフォンヴィレブランド因子(von Willebrand factor:vWF)のみを切断します。vWFは、もともと血管内皮細胞でUL-vWFM(unusually large-vWF multimer)という大きな塊で産生され、ADAMTS13により細かく切断されて適切な大きさのvWFとなることで、適切な止血作用を持ちます。しかし、ADAMTS13の機能が大きく低下してしまうと、不適切に大きいvWFが残ってしまい、余分な止血作用を発揮し血小板血栓を血管内に生じてしまいます。vWFによる血栓は、血管が細ければ細いほどできやすいため、微小血管内で血小板血栓が生じ、TMAを発症すると考えられています。
このADAMTS13の活性の低下によるTMAでは、1)腎機能障害は比較的軽度で、2)発熱、3)精神障害が強く出る血栓性血小板減少性紫斑病(TTP)となることが知られています。4)血小板減少、5)溶血性貧血と合わせてTTPの5徴と呼ばれます。ただし、全ての症状が揃うことは30%程度と報告されています。
ADAMTS13の活性が大きく低下する原因としては、先天的な遺伝子異常と、後天的な要因があります。ADAMTS13の先天的な遺伝子異常によりTMAを発症する疾患はUpshaw-Schulman症候群と呼ばれています。生まれたばかりの時から重症の黄疸と血小板減少を生じるまれな疾患です。ADAMTS13の後天的な活性低下の原因としては、ADAMTS13に対する自己抗体(IgG型)が最も重要です。基礎疾患がないものを原発性、ほかの疾患によって発症する場合には二次性や続発性などと呼びます。乳幼児から高齢者まで幅広く発症し、男女比は1:2とやや女性に多いといわれています。有病率は人口100万人に4人程度と報告されてきましたが、近年の診断技術の向上のため、最近ではもっと多いものと推測されています。その他、抗血小板薬であるチクロピジンやクロピトグレルが原因となることも知られつつあり、これらを内服している患者さんでは定期的な血液検査で早期発見を行っております。
ADAMTS13活性の測定はTMAの病勢の評価に有用ですが、ADAMTS13活性の低下がないこともあり、この検査のみではTMAの除外はできないことに注意が必要です。また、医療機関によってはADAMTS13活性の測定が難しい場合や結果が判明するまで時間がかかる場合もあり、TTPを診断するためにPLASMICスコアやFrenchスコアの使用も推奨されています。血栓性血小板減少性紫斑病(TTP)の名称で指定難病に登録されており、中等症以上であれば医療費助成の対象になっています。
4. コバラミン代謝異常
先天的なコバラミン代謝異常で起こる高ホモシステイン血症が、TMAの病態を生じることがあります。非常に重症で新生児・乳児期に死に至るまれな疾患です。
5. 薬剤性(例:キニン)
かつて抗マラリア薬として用いられたキニンによりTMAの病態をきたすことが知られています。キニンは副作用が多いため、近年ではほかの抗マラリア薬が使用されます。また、その他の薬剤が原因でTMAが起きる報告もありますが、薬剤性TMAでは被疑薬を中止すれば自然軽快することも多いです。
病因ははっきりしないが他疾患との関連が知られているTMA
1. HIV感染
HIVのウイルス自体が直接血管内皮細胞を障害することで、TMAの症状を生じることがあります。ただしまれです。
2. 悪性腫瘍
播種を伴う胃がん、大腸がん、前立腺がんなどの患者さんにTMAを発症することがあります。胃がんや乳がんで使用されるマイトマイシンもTMAに関わることがあります。
3. 臓器移植、造血幹細胞移植
腎移植や肝移植、造血幹細胞移植後にTMAを発症することがあります。移植前に大量に投与される抗がん剤の影響や、移植後に使用されるカルシニューリン阻害薬という免疫抑制薬が関与していると考えられています。ADAMTS13の作用は低下しておらず、微小血管の内皮細胞障害が原因と考えられていますが、未だ詳しいメカニズムは不明です。
4. 膠原病
全身性エリテマトーデスの2~8%程度にTMAが合併するといわれています。メカニズムは不明な点もありますが、ADAMTS13に対する自己抗体や、血小板に発現するCD36という糖蛋白に対する自己抗体が産生されることで、微小血管の内皮細胞障害が生じTMAを発症するものと推察されています。全身性エリテマトーデスの患者さんに血小板減少症が生じた場合には様々な原因を想定して検査を進めますが、その中でTMAの可能性を忘れないようにしなければなりません。さらにTMAの症状の一つとして精神症状がありますが、全身性エリテマトーデス自体でも精神症状が出る可能性があります。全身性エリテマトーデスの場合には精神症状の変動が少なくだんだん悪くなっていく傾向にありますが、TMAによる精神症状が時間帯によって大きく変動することが特徴なので、両者を区別することができます。ADAMTS13活性は低下することもあれば正常の場合もあります。
また、強皮症にも合併しやすいことが知られています。強皮症腎クリーゼの50~70%以上においてTMAが併発し、TMAが先行することもあります。その他、抗リン脂質抗体症候群、多発性筋炎/皮膚筋炎などでもまれですがTMAを合併することがあります。
膠原病に合併するTMAにおいて、ADAMTS13活性の著減(0.5%未満)例は5~23%程度であり、原因が不明のTMAにおいては70%程度で同活性が著減していたことと比べて、低率であることが特徴です。
5. 妊娠
妊娠とTMAとの関係はあきらかでありません。ただ、妊娠後期に発症する子癇やHELLP症候群はTMAに類似した病状になることがあります。
6. 糸球体腎炎
ある種の糸球体腎炎とTMAが合併することもあります。いずれも補体が関与しているものと考えられています。
7. その他の遺伝疾患
8. 分類不能
TMAとの鑑別が必要な別の病態
TMAの診断のためには、ほかの疾患の可能性を否定しなければなりません。血小板が減少するほかの病態として、播種性血管内凝固症候群(DIC)、免疫性血小板減少症(ITP)、血球貪食症候群(HPS)、薬剤などによる骨髄低形成、劇症型抗リン脂質抗体症候群(CAPS)、ヘパリン起因性血小板減少症、自己免疫性溶血性貧血、悪性貧血、サイトメガロウイルスなどの感染症などがあり、これらとの区別が重要になります(表1)。
表1.TMAとの鑑別が必要な病態
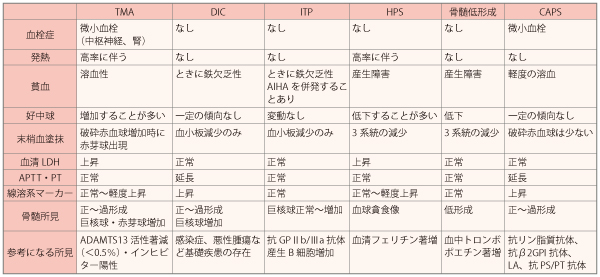
DIC:播種性血管内血液凝固症、ITP:免疫性血小板減少症、HPS:血球貪食症候群、CAPS:劇症型抗リン脂質抗体症候群、LA:ループスアンチコアグラント、PS/PT:ホスファチジルセリン依存性プロトロンビン
治療
TMAの治療は、その原因によって異なりますが、基本は血漿交換療法になります。血漿交換は新鮮凍結血漿(FFP)の補充(50~80 mL/kg/日)を3~5日連続して行います。血小板数や腎機能をみながら実施間隔を調整します。著効例では血漿交換を1~3回実施すると効果がみられますが、効果判定のために血漿交換を10回程度まで実施することが多いです。
腸管出血性大腸菌によるTMAの場合
基本的には自然に治癒するため、症状に合わせた治療を行います。腎機能障害が重度の場合には一時的な血液透析を行うことがあります。治療成績は良好です。
補体介在性TMAの場合
血漿交換療法に加えて、抗C5抗体薬であるエクリズマブ(ソリリス®)、ラブリズマブ(ユルトミリス®)が日本でも保険適用となりました。これらは補体C5に結合することにより、補体の異常な活性化を抑制します。抗C5抗体薬投与を行う際は、髄膜炎菌感染のリスクが高まりますので、髄膜炎菌ワクチンの接種や予防的抗菌薬の投与を行います。その他に淋菌、肺炎球菌、インフルエンザ桿菌の感染リスクも報告されています。抗C5抗体薬は病気が落ち着いてからも、しばらくの間は投与を継続することがあります。
ADAMTS13の先天的な遺伝子異常によるTMA(Upshaw-Schulman症候群)の場合
ADAMTS13を補充するために2~3週間おきに新鮮凍結血漿の輸注が必要になります。
ADAMTS13の後天的な異常によるTMAの場合
かつては極めて重症で難治性の病気でしたが、血漿交換という治療の有効性が確立してからは治療成績が大きく向上しています。ステロイドが併用されることもあります。
膠原病に伴うTMAの場合
血漿交換の有効性が知られるようになってからは治療成績が向上しています。ADAMTS13活性が著減(0.5%未満)し、抗ADAMTS13抗体(インヒビター)が陽性の場合(定型的TMA)には、血漿交換療法に加えて、自己免疫を抑制するためにステロイド大量療法を追加します。治療抵抗(薬物がききにくい)例や再発例にはシクロホスファミド(商品名:エンドキサン®)間欠静注療法やシクロスポリン(商品名:ネオーラル®)などの免疫抑制薬を併用します。2020年2月より抗CD20キメラ抗体のリツキシマブ(商品名:リツキサン®)が再発・難治の後天性TTPに対して保険適応になりました。さらに、最近では、vWFに対する抗体製剤であるカプラシズマブ(商品名:カブリビ®)が急性期の重篤な血栓症を予防するために使用できるようになりました。カプラシズマブは血小板とvWFの結合を阻害することで血栓の形成を抑制しますが、上記の治療と併用する必要があります。ADAMTS13活性が著減する例ではインヒビター陽性になることが多く、血小板減少が高度である一方で腎障害は少ないのが特徴です。
一方、ADAMTS13活性が著減していない(0.5%以上)例(非定型TMA)では、ステロイドや免疫抑制薬による免疫抑制療法の必要性について議論が分かれていますが、血漿交換のみで改善が乏しい例や再発例には、免疫抑制療法が実施されることが多いです。ただし、強皮症に合併したTMAでは免疫抑制療法で改善が乏しく予後(病気の見通し)が悪くなることがあります。この場合、免疫グロブリン大量療法や脾摘が試され有効との報告もありますが、効果が無い場合もあります。非定型TMAでは腎障害が多くみられ、予後が悪い傾向があります。
強皮症の腎クリーゼに伴うTMAに対する治療は原則不要で、アンギオテンシン変換酵素阻害薬の投与によって血圧のコントロールができればTMAも改善することが多いです。
臓器移植、造血幹細胞移植に伴うTMAの場合
血栓性血小板減少性紫斑病(TTP)で有効な血漿交換は、移植後のTMAに対して効果的でありません。治療方法は現時点で確立されておらず、予後は極めて不良です。
なお、TMAは血小板が減少する病気なので血小板を補充すればよいと思われがちですが、血小板輸血は原則禁忌になります。なぜなら、新しく補充された血小板が、血管内の血栓をさらに増大させてしまい症状が悪くなってしまうからです。ただし、出血傾向が非常に強い場合や侵襲的処置が必要な場合には、血漿交換直後に血小板を注意して投与する場合もあります。
また、アスピリンなどの抗血小板剤の効果については不明ですが、急性期には無効で、治った後の再発予防に用いることが多いです。
生活上の注意
腸管出血性大腸菌による腸炎を発症した場合には4~10日にTMAの症状が出現する可能性があります。紫斑が出現したり、鼻血や歯グキからの出血が増えたりした場合には、速やかに医療機関を受診してください。TMAを発症したら基本的に入院治療になります。場合によっては再発することがあるため、退院された後に同様の症状が出てきた場合には速やかに受診するようにしてください。
慶應義塾大学病院での取り組み
上記のようにTMAは様々な原因で生じます。各科の医師がTMAの早期発見に努め、破砕赤血球やASAMTS13活性などの検査を積極的に施行しております。血漿交換療法や免疫抑制療法を含めて適切な治療を選択し治療効果の向上に努めています。
さらに詳しく知りたい方へ
- 難病情報センター

血栓性血小板減少性紫斑病(TTP)、非典型溶血性尿毒症症候群の各疾患名で検索してください。 - 腸管出血性大腸菌Q&A(厚生労働省)
- 慶應義塾大学医学部リウマチ・膠原病内科
SHARE