先天性副腎低形成症とは?
先天性副腎低形成症は、先天的な原因により副腎と呼ばれる内分泌器官が十分に形成されず、生命活動の維持に必須な副腎皮質ホルモン(糖質コルチコイド、鉱質コルチコイドなど)が欠乏する疾患です。国が定める指定難病であり、わが国における患者数は約1,000人と推定されています。これまでの研究から、副腎細胞の発生や増殖に関わる遺伝子の異常が先天性副腎低形成症の原因となることが知られていましたが、約30%の患者さんは原因不明でした。
次世代遺伝子解析技術を用いた病因究明
今回、我々の研究チームは、原因不明の先天性副腎低形成症患者さん24人の遺伝子を次世代遺伝子解析技術などで分析しました。その結果、11人がSAMD9遺伝子に異常を持つことをつきとめることに成功しました。SAMD9遺伝子異常を持つ11人は、先天性副腎低形成症以外にも造血異常、易感染性(感染症にかかりやすくなること)、成長障害、性腺症状(精巣や卵巣の異常)、消化器症状が共通していました。このような組み合わせをとる疾患はこれまで世界的に報告がなく、まったく新しい疾患単位であることが判明しました。研究チームは6つの主症状を表す英語の頭文字をとり、疾患名を「MIRAGE(ミラージュ)症候群」とすることを提唱しました(図1)。この病名は2016年7月25日づけで世界的な遺伝子疾患のオンラインカタログOMIM![]() に登録されました。
に登録されました。

図1
MIRAGE症候群は6つの症状(造血異常M、易感染性I、成長障害R、先天性副腎低形成症A、性腺症状G、消化器症状E)の頭文字を意味する。その原因はSAMD9遺伝子の働きが異常に活性化されることである。
研究チームはMIRAGE症候群のメカニズムを調べるため、培養細胞を用いた実験を行いました。培養細胞で正常SAMD9遺伝子を働かせると、細胞増殖がわずかに遅くなりましたが、異常SAMD9遺伝子を働かせると、細胞増殖が強力に抑えられることがわかりました。また、これまでの研究からSAMD9分子がエンドソーム(注1)系の働きを調節する可能性が示されていたことを参考に、患者さんの細胞を詳細に分析したところ、後期エンドソームの特徴を持つ巨大な小胞が細胞内に充満していることを明らかにしました。SAMD9遺伝子異常が後期エンドソームの形成過剰を引き起こし、その結果、細胞の状態が不良となっていると考えられました。
骨髄異形成症候群(MDS)との関わり
MIRAGE症候群の患者さん11人のうち2人は、子どもでは極めて稀な血液疾患、骨髄異形成症候群(MDS)を発症していました。2人いずれにおいてもMDS骨髄細胞に7番染色体欠失(通常2本ある7番染色体のうち1本が失われる現象)が起きていました。SAMD9遺伝子が7番染色体上に位置することに着目し、2人のMDS骨髄細胞のSAMD9遺伝子を調べたところ、遺伝子異常を示すシグナルが大幅に減少しており、2本ある7番染色体のうち異常SAMD9遺伝子が存在する染色体が欠失したことがわかりました(図2)。
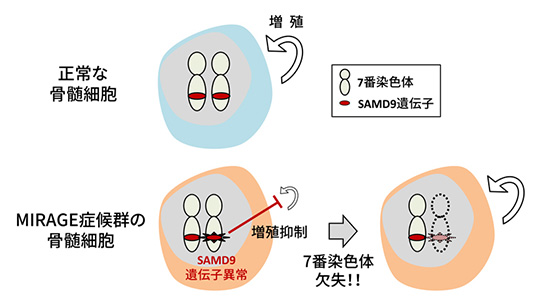
図2.MIRAGE症候群の患者さんの骨髄細胞における7番染色体欠失
染色体異常はMDS、白血病に限らず様々な腫瘍でみられ、細胞の腫瘍化に関わると想定されています。染色体は「生命の設計図」とも言える重要な構造であり、染色体異常が生じた細胞の大部分は腫瘍化することなく死滅しますが、なんらかの理由で染色体異常細胞が正常細胞よりも増殖しやすくなると、次第に腫瘍を形成すると考えられます。今回、MIRAGE症候群の患者さんでみられた7番染色体欠失は、細胞増殖抑制作用を持つ異常SAMD9遺伝子を除去し、細胞増殖速度を高める効果を与えたと推測されます。一方、異常SAMD9遺伝子を除去する代償として、7番染色体上に位置する1,000個以上の遺伝子が失われ、この欠失がMDS発症の引き金になったと考えられます。
今後の展望
MIRAGE症候群は多彩な全身症状を示す疾患であり、副腎細胞、骨髄細胞、消化器細胞、免疫細胞などでSAMD9分子が働いていることが推測されます。今後、SAMD9分子の働きを詳しく調べることにより、これらの臓器の疾患の新しい診断法、治療法の開発に役立てられると考えられます。
【用語解説】
注1)エンドソーム
エンドソームとはさまざまの物質を細胞の外側から内側に取り込んだり、内側から外側に放出するときに働く細胞内の小器官です。
参考文献
SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7.
Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, Toyoshima K, Tanaka Y, Fukuzawa R, Miyako K, Kinjo S, Ohga S, Ihara K, Inoue H, Kinjo T, Hara T, Kohno M, Yamada S, Urano H, Kitagawa Y, Tsugawa K, Higa A, Miyawaki M, Okutani T, Kizaki Z, Hamada H, Kihara M, Shiga K, Yamaguchi T, Kenmochi M, Kitajima H, Fukami M, Shimizu A, Kudoh J, Shibata S, Okano H, Miyake N, Matsumoto N, Hasegawa T.
Nat Genet. 2016 Jul;48(7):792-7. doi: 10.1038/ng.3569. Epub 2016 May 16.
http://www.nature.com/ng/journal/v48/n7/index.html

左から石井智弘(小児科学教室専任講師)、鳴海覚志(論文執筆時:小児科学教室 特任助教、現国立成育医療研究センター)、筆者、天野直子(小児科学教室共同研究員)
最終更新日:2016年11月1日
記事作成日:2016年11月1日